









W modelowaniu molekularnym spotykamy różne formaty zapisu struktury związków, najpopularniejszymi formatami są układ kartezjański x,y,z, z-macierz, .pdb, czy .cif. Prawda jest taka, że istnieje cały szereg formatów zapisu, medot konwersji, rodzaju przydatności czy oprogramowania.
Dlaczego tak dużo formatów zapisów struktur do modelowania molekularnego?
Tak jak w muzyce spotyka się całą gamę plików np.: ogg, mp3, mp4, acc, także i w chemii molekularnej spotyka się. Spowodowane jest to wygodą przedstawiania struktury oraz danych i tak np.: cif – zoptymalizowany jest do przechowywania informacji o strukturze krystalograficznej, a pdb do przechowywania informacji o białkach. Formaty różne są tak opracowywane aby najbardziej odpowiadały oczekiwaniom producentów oprogramowania – optymalizacja wydajności i przechowywania danych także i tych dodatkowych
Jakie są formaty zapisów struktur do modelowania molekularnego?
Tak jak powyżej wspomnieliśmy jest cała gama formatów (.log, .inp, gjf, .acc, .png2, .gzmat, .pdb, .cube, .fchk, .chk, . Jin, .k, .mol, .mol2, .ml2 etc...)
Jak przekonwertować (zamienić jeden format zapisu na drugi format zapisu)?
Jeżeli zdarzy się problem czytania jednego formatu przez program wówczas najlepszym sposobem jest zmiana formatu. Do tego celu można skorzystać z różnych programów, ale najlepszym rozwiązaniem jest skorzystanie z programu OPEN BABEL który oferuję w prosty sposób konwersję na różne formaty. Po uruchomieniu tego oprogramowania widzimy dwa główne okna, w jednym oknie (prawym) wklejamy strukturę którą chcemy przekonwertować, ustawiamy bieżący format, a następnie w drugim oknie wybieramy format docelowy. I tak struktura etanolu – możemy przedstawić w postaci:
======================================
.mol2, .ml2 – Sybyl
# STRUKTURA czasteczki
# Etanol
#
#
@<TRIPOS>MOLECULE
Molecule Name
9 8
SMALL
NO_CHARGES
@<TRIPOS>ATOM
1 C1 -0.5134 0.5357 0.0000 C
2 H2 -0.1567 -0.4731 0.0000 H
3 H3 -0.1567 1.0401 -0.8737 H
4 H4 -1.5834 0.5357 0.0000 H
5 C5 -0.0001 1.2617 1.2574 C
6 H6 -0.3551 2.2710 1.2564 H
7 H7 -0.3583 0.7584 2.1311 H
8 O8 1.4299 1.2594 1.2587 O
9 H9 1.7504 1.7119 2.0424 H
@<TRIPOS>BOND
1 1 2 1
2 1 3 1
3 1 4 1
4 1 5 1
5 5 6 1
6 5 7 1
7 5 8 1
8 8 9 1
======================================
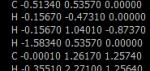
.xyz-układ kartezjański
9
Molecule Name
C -0.51340 0.53570 0.00000
H -0.15670 -0.47310 0.00000
H -0.15670 1.04010 -0.87370
H -1.58340 0.53570 0.00000
C -0.00010 1.26170 1.25740
H -0.35510 2.27100 1.25640
H -0.35830 0.75840 2.13110
O 1.42990 1.25940 1.25870
H 1.75040 1.71190 2.04240
======================================
.tmt – turbomole coordinate format
$coord
-0.97018539257171 1.01232628515907 0.00000000000000 c
-0.29612008378650 -0.89402942973447 0.00000000000000 h
-0.29612008378650 1.96550414260583 -1.65105371540690 h
-2.99219234631485 1.01232628515907 0.00000000000000 h
-0.00018897261250 2.38426745190441 2.37614162956694 c
-0.67104174698522 4.29156802986044 2.37425190344195 h
-0.67708887058520 1.43316829319514 4.02719534497384 h
2.70211938612833 2.37992108181693 2.37859827352943 o
3.30777660918878 3.23502215337653 3.85957663768691 h
$end
======================================
.pdb – Protein databank
COMPND Molecule Name
AUTHOR GENERATED BY OPEN BABEL 2.3.2
HETATM 1 C LIG 1 -0.513 0.536 0.000 1.00 0.00 C
HETATM 2 H LIG 1 -0.157 -0.473 0.000 1.00 0.00 H
HETATM 3 H LIG 1 -0.157 1.040 -0.874 1.00 0.00 H
HETATM 4 H LIG 1 -1.583 0.536 0.000 1.00 0.00 H
HETATM 5 C LIG 1 -0.000 1.262 1.257 1.00 0.00 C
HETATM 6 H LIG 1 -0.355 2.271 1.256 1.00 0.00 H
HETATM 7 H LIG 1 -0.358 0.758 2.131 1.00 0.00 H
HETATM 8 O LIG 1 1.430 1.259 1.259 1.00 0.00 O
HETATM 9 H LIG 1 1.750 1.712 2.042 1.00 0.00 H
CONECT 1 2 3 4 5
CONECT 1
CONECT 2 1
CONECT 3 1
CONECT 4 1
CONECT 5 1 6 7 8
CONECT 5
CONECT 6 5
CONECT 7 5
CONECT 8 5 9
CONECT 9 8
MASTER 0 0 0 0 0 0 0 0 9 0 9 0
END
======================================
.adf – ADF cartesian input format
TITLE Molecule Name
CHARGE 0 0
Number of atoms
9
ATOMS Cartesian
C -0.51340 0.53570 0.00000
H -0.15670 -0.47310 0.00000
H -0.15670 1.04010 -0.87370
H -1.58340 0.53570 0.00000
C -0.00010 1.26170 1.25740
H -0.35510 2.27100 1.25640
H -0.35830 0.75840 2.13110
O 1.42990 1.25940 1.25870
H 1.75040 1.71190 2.04240
End
Basis
End
Geometry
End
======================================
.cif – crystalografic data format
# CIF file generated by openbabel 2.3.2, see http://openbabel.sf.net
data_I
_chemical_name_common 'Molecule Name'
loop_
_atom_site_type_symbol
_atom_site_label
_atom_site_Cartn_x
_atom_site_Cartn_y
_atom_site_Cartn_z
C C1 -0.51340 0.53570 0.00000
H H2 -0.15670 -0.47310 0.00000
H H3 -0.15670 1.04010 -0.87370
H H4 -1.58340 0.53570 0.00000
C C5 -0.00010 1.26170 1.25740
H H6 -0.35510 2.27100 1.25640
H H7 -0.35830 0.75840 2.13110
O O8 1.42990 1.25940 1.25870
H H9 1.75040 1.71190 2.04240
======================================
.gamin – GAMMES format
$CONTRL COORD=CART UNITS=ANGS $END
$DATA
Molecule Name
C1
C 6.0 -0.5134000000 0.5357000000 0.0000000000
H 1.0 -0.1567000000 -0.4731000000 0.0000000000
H 1.0 -0.1567000000 1.0401000000 -0.8737000000
H 1.0 -1.5834000000 0.5357000000 0.0000000000
C 6.0 -0.0001000000 1.2617000000 1.2574000000
H 1.0 -0.3551000000 2.2710000000 1.2564000000
H 1.0 -0.3583000000 0.7584000000 2.1311000000
O 8.0 1.4299000000 1.2594000000 1.2587000000
H 1.0 1.7504000000 1.7119000000 2.0424000000
$END
======================================
.gzmat, .zmat – z-macierz dla plików Gaussiana
#Put Keywords Here, check Charge and Multiplicity.
Molecule Name
0 1
C
H 1 r2
H 1 r3 2 a3
H 1 r4 2 a4 3 d4
C 1 r5 2 a5 3 d5
H 5 r6 1 a6 2 d6
H 5 r7 1 a7 2 d7
O 5 r8 1 a8 2 d8
H 8 r9 5 a9 1 d9
Variables:
r2= 1.0700
r3= 1.0700
a3= 109.47
r4= 1.0700
a4= 109.47
d4= 120.00
r5= 1.5400
a5= 109.47
d5= 240.00
r6= 1.0699
a6= 109.47
d6= 179.88
r7= 1.0700
a7= 109.47
d7= 299.89
r8= 1.4300
a8= 109.47
d8= 59.89
r9= 0.9600
a9= 109.50
d9= 179.97
Podsumowanie formatów zapisów struktur
Jak widzimy istnieje naprawdę wiele formatów plików, które różnią się budową pliku oraz sposobem zapisu. Te kilka przykładów struktury etanolu przekonwertowanej na różne formaty świadczą o tym. Konwersje można przeprowadzać programem openbabel lub obabel strona: openbabel.org.