









Aktualności
NBO -2009
Analiza NBO (ang. Natura Bond Orbital)
Omówienie analizy NBO na przykładzie cząsteczki wody
Zapis cząsteczki w postaci Z-macierzy i w układzie kartezjańskim

Struktura cząsteczki jest zapisywana przeważnie w formacie macierzy-Z (ang. z-matrix) z wykorzystaniem współrzędnych wewnętrznych lub w układzie kartezjańskim.
Skrypt do Chemii Kwantowej - Dr Łukasz Rajchel
Drugim doskonałym kompendium wiedzy z chemii kwantowej która jest dostępna w internecie jest skrypt doktora Łukasz Rajchela z Pracowni Chemii Kwantowej Wydziału Chemii Uniwersytetu Warszawskiego. -obecnie Faculty of Chemistry, University of Duisburg-Essen, Germany
Energia - Geometria - Optymalizacja
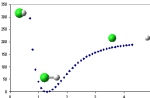 Mając najprostszą cząsteczkę dwuatomową wiemy, że istnieje pewna optymalna odległość między atomami gdzie energia układu jest najmniejsza. Odległość tą nazywamy długością wiązania. Możemy ją obliczyć poprzez obliczanie energii dla kolejnych odległości między atomami. W ten sposób możemy sporządzić wykres zależności energii od odległości między atomami. W takim przypadku uzyskaną krzywą nazywamy krzywą energii potencjalnej. Minimum energii odpowiada geometrii, w której cząsteczka występuje w stanie stacjonarnym, to znaczy w badanym medium ilość cząsteczek o danej geometrii jest największa.
Mając najprostszą cząsteczkę dwuatomową wiemy, że istnieje pewna optymalna odległość między atomami gdzie energia układu jest najmniejsza. Odległość tą nazywamy długością wiązania. Możemy ją obliczyć poprzez obliczanie energii dla kolejnych odległości między atomami. W ten sposób możemy sporządzić wykres zależności energii od odległości między atomami. W takim przypadku uzyskaną krzywą nazywamy krzywą energii potencjalnej. Minimum energii odpowiada geometrii, w której cząsteczka występuje w stanie stacjonarnym, to znaczy w badanym medium ilość cząsteczek o danej geometrii jest największa.
Przeliczanie energii
 Jednym z podstawowych umiejętności podczas interpretacji obliczeń teoretycznych jest przeliczanie wartości energii na różne jednostki. Obliczenia wykonywane na przykład programem Gaussian zwracają wartość energii w Hartree.
Jednym z podstawowych umiejętności podczas interpretacji obliczeń teoretycznych jest przeliczanie wartości energii na różne jednostki. Obliczenia wykonywane na przykład programem Gaussian zwracają wartość energii w Hartree.
Różne formaty zapisów struktury
W modelowaniu molekularnym spotykamy różne formaty zapisu struktury związków, najpopularniejszymi formatami są układ kartezjański x,y,z, z-macierz, .pdb, czy .cif. Prawda jest taka, że istnieje cały szereg formatów zapisu, medot konwersji, rodzaju przydatności czy oprogramowania.
Wstęp do modelowania
Modelowanie Komputerowe
Modelowanie molekularne, zwane inaczej chemią obliczeniową, ma zastosowanie zarówno w chemii, biochemii, inżynierii materiałowej jak i w nanotechnologii. Rozwój komputerów oraz programów sprawił, że w ostatnich czasach stało się ono dostępne dla wszystkich, a obliczenia chemiczne można już przeprowadzać na komputerze osobistym. Koszt oraz czas badań teoretycznych jest niejednokrotnie o wiele mniejszy niż wstępne badania praktyczne a uzyskane wyniki coraz bardziej zbliżają się do wyników eksperymentalnych. Korzystając z dobrodziejstw chemii teoretycznej możemy już dziś z bardzo dobrymi rezultatami przewidzieć właściwości fizyczne i chemiczne, modelować reakcje oraz stany przejściowe, symulować widma spektroskopowe, czy też badać układy bardziej skomplikowane, jakim są centra aktywne enzymów. Możemy także projektować leki oraz nowe materiały. Modelowanie komputerowe często nie może dać jednoznacznej odpowiedzi na zadawane pytania, ale w znaczny sposób pomaga zrozumieć oraz rozwiązać problemy. Wszechstronnie wykształcony chemik powinien umieć korzystać z najnowszych dobrodziejstw, jakim niewątpliwie jest wsparcie badań eksperymentalnych obliczeniami.
Elektryczny moment dipolowy
 Elektryczny moment dipolowy jest wektorową wielkością fizyczną, która opisuje dipol. Dipol jest układem ładunków elektrycznych o tej samej wartości lecz posiadających przeciwny znak ... więcej w artykule Pawła Grabowskiego.
Elektryczny moment dipolowy jest wektorową wielkością fizyczną, która opisuje dipol. Dipol jest układem ładunków elektrycznych o tej samej wartości lecz posiadających przeciwny znak ... więcej w artykule Pawła Grabowskiego.
Potencjał korelacyjno-wymienny B3LYP
Hyperchem - jeszcze raz od podstaw
Mamy zaszczyt przedstawić fragment kursu autorstwa dr Piotra Wojciechowskiego z Politechniki Wrocławskiej. Fragment kursu opisuje pierwsze kroki w programie chemicznych HyperchemR. Oryginalna strona kursu znajduje się http://eportal-ch.pwr.wroc.pl Jeszcze raz dziękujemy autorowi za udostępnienie materiałów.
Struktura związków w przestrzeni X,Y,Z
Ćwiczenie
Struktura związków w przestrzeni X,Y,Z
Jednym ze sposobów zapisu struktury jest podanie współrzędnych kolejnych atomów w postaci położenia x,y,z – korzystając z poniższych danych przeprowadź wizualizacje podanych cząsteczek. Powinieneś otrzymać podobne wyniki jak przedstawiamy w galerii.
Amarant (E123) – barwnik organiczny używany jako barwnik spożywczy. Ma barwę ciemnoczerwoną oraz dobrą wytrzymałość na wysoką temperaturę i światło. Obecnie stosowany do barwienia nielicznych produktów spoŜywczych, takich jak: niskoprocentowe napoje alkoholowe oraz kawior, a także to barwienia produktów takich jak szminki.
Testowanie różnych metod i baz do obliczeń geometrii
Ćwiczenie
Testowanie różnych metod i baz do obliczeń geometrii
Obliczona geometria cząsteczki zależy od wybranej metody i bazy. Używając lepszych metod oraz baz, mamy większe prawdopodobieństwo, że obliczone parametry geometryczne zbliżą się do wartości eksperymentalnych, które wyznaczone są zawsze z pewnym błędem. Większa baza i dokładniejsza metoda pociągają za sobą większe wymagania sprzętowe. Dlatego zawsze wybierając metodę i bazę na podstawie studiów literaturowych z określonej dziedziny, w której chcemy prowadzić obliczenia, musimy oszacować, na jak dokładną metodę możemy sobie pozwolić.
Aby samemu zbadać, jaką różnice w parametrach geometrycznych (długość wiązań, kąty) dają różne metody przeprowadź optymalizację cząsteczki aldehydu mrówkowego H2C=O, oraz porównaj z wartościami eksperymentalnymi.[1] Obliczenia przeprowadź:
I za pomocą mechaniki kwantowej metodą Amber
II jedną z wybranych metod półempirycznych:
a) AM1,
b) PM3,
c) PM3MM,
d) CNDO,
e) INDO,
III metodami ab inito: HF, MP2 oraz metodami DFT: B3LYP, B3PW91 w trzech wybranych bazach:
a) STO-3G,
b) 3-21G,
c) 6-31G(d) równoważne w zapisie starej notacji 6-31G*,
d) 6-31G(d,p) równoważne w zapisie starej notacji 6-31G**,
e) 6-31+G(d,p) równoważne w zapisie starej notacji 6-31+G**,
f)* aug-pvdz,
Otrzymane wyniki porównaj z wartościami eksperymentalnymi wiedząc, że cząsteczka jest płaska oraz kąt tworzony przez atomy HCH wynosi 116.5°, długość wiązania C-H 1.116 Å, C=O 1.208 Å.
* Dodatkowo dla struktur z optymalizowanych metodami AM1 oraz B3LYP/6-31G(d) przeprowadź optymalizację metodą B3LYP/6-31G(d,p), porównaj czasy obliczeń. Od czego zależy czas przeprowadzonej optymalizacji?
[1] Dallos, M.; Müller, T.; Lischka, H.; Shepard, R., Geometry optimization of excited valence states of formaldehyde using analytical multireference configuration interaction singles and doubles and multireference averaged quadratic coupled-cluster gradients, and the conical intersection formed by the 1 1B1(σ-π*) and 2 1A1(π-π*) states. J. Chem. Phys. 2001, 114, (2), 746-757.
Analiza konformacyjna 1,3-difluoropropanu
Ćwiczenie
Analiza konformacyjna 1,3-difluoropropanu
Zmień wartości poszczególnych kątów dwuściennych cząsteczki 1,3-difluoropropanu. Następnie przeprowadź optymalizację. Znajdź wszystkie struktury w minimach
energetycznych poprzez startowanie z różnych konformerów.

Określ minimum globalne. Obliczenia przeprowadź metodą B3LYP w bazie 6-31G(d) oraz w metodzie PM3. Bazując na lepszej metodzie B3LYP, porównaj z wynikami uzyskanymi metodą PM3.
Badanie struktury przestrzennej hemoglobiny1 - posługiwanie się bazą PDB
![]() Ćwiczenie
Ćwiczenie
Badanie struktury przestrzennej hemoglobiny1 - posługiwanie się bazą PDB
Podczas modelowania układów biologicznych opartych na białkach pierwszym etapem jest uzyskanie struktury przestrzennej białka. Geometrię białka uzyskuje się z badań eksperymentalnych głównie z rentgenografii strukturalnej lub z badań techniką nuklearnego rezonansu magnetycznego (NMR). Białka o ustalonej już geometrii są zdeponowane w różnych bazach. Jedną z takich baz oferująca ok. 30 tysięcy struktur jest PDB (Protein Data Bank) dostępna pod adresem: http://www.rcsb.org/pdb/.
Po wejściu na stronę www.rscb.org/pdb/, wyszukiwanie odpowiedniej struktury zaczynamy od wpisania słów kluczowych. Wyszukiwanie hemoglobiny dokonujemy wpisując w polu Serach the Archive angielską nazwę: hemoglobin. Z uzyskanych rezultatów wybieramy odpowiednie białko np.:

Klikając na odpowiedni link (EXPLORE, lub numer 1BZ0), uzyskujemy więcej informacji o białku. Spakowaną strukturę białka pobieramy klikając ikonę . Po pobraniu struktury i jej rozpakowaniu, otwieramy ją odpowiednim programem. Struktura białka została ustalona metodami rentgenograficznymi związku z czym nie zawiera atomów wodoru, dlatego potrzebne jest uzupełnienie atomami wodoru brakujących miejsc.
G
Posługując się programem GaussView wybieramy z menu File opcję Open?, a następnie ustawiamy w opcji Pliki Typu: Brookhaven PDB Files (*.pdb *.ent) i otwieramy rozpakowany plik ze strukturą białka. Program, podczas ładowania struktury, proponuje dodanie atomów wodoru (Would you like to add hydrogens to your PDB structure?), zgadzamy się wybierając Yes. Po tych operacjach możemy przystąpić do analizowania struktury i odpowiedzi na pytania.
HyperChem
Posługując się programem HyperChem, wybieramy z menu File opcję Open, a następnie ustawiając w opcji Pliki typu: Brookhaven PDB (*.ENT), otwieramy rozpakowany plik ze strukturą białka. Uzupełnienie wodorami dokonujemy wybierając z menu Build opcję Add Hydrogens. Po tych operacjach możemy przystąpić do analizowania struktury i odpowiedzi na pytania.
Rasmol
Dodatkowe informacje oraz podejrzenie struktury drugorzędowej białka możemy dokonać, korzystając z darmowego programu RasMol.2 Otwieramy plik ze strukturą, wybierając z menu File opcję Open. Zmianę sposobu wyświetlania struktury dokonujemy, korzystając z menu Display wybierając odpowiednią opcje (np.: Ribbons lub Wireframe), zaś zmianę kolorów elementów wyświetlanych dokonujemy wybierając którąś z opcję (np.: Chain) z menu Colours.
Oglądając i analizując strukturę hemoglobiny odpowiedz na następujące pytania:
- Z ilu podjednostek białkowych składa się hemoglobina?
- Jaka jest struktura szkieletu hemu -płaska czy wypukła?
- Z czym koordynują atomy żelaza występujące w białku (jakie to są aminokwasy, grupy)?
- Jakie struktury drugorzędowe występują w białku? -proszę skorzystać z programu RasMol.
[1] Ze względu na dużą ilość atomów w cząsteczce dokonujemy jedynie w tym ćwiczeniu analizy gotowej struktury białka po uzupełnieniu jej atomami wodoru.
[2] Więcej informacji oraz program można uzyskać na stronach: http://www.bernstein-plus-sons.com/software/rasmol/doc/rasmol.html
Ustalanie bezwzględnej konfiguracji R, S na podstawie wzorów przestrzennych
Ćwiczenie
Ustalanie bezwzględnej konfiguracji R, S na podstawie wzorów przestrzennych
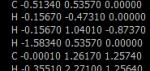
Narysuj jedną z wybranych struktur1 lub pobierz strukturę w postaci pliku xyz która jest umieszczona na koncu dokumnetu. Zoptymalizuj układ metodą półempiryczną PM3. Posługując się modelem trójwymiarowym określ konfigurację R, S chiralnych atomów węgla, poprzez obracanie cząsteczki w odpowiednich kierunkach.
Opis określania konfiguracji R, S - zgodnej z konwencją Cahna-Ingolda-Preloga znajdziemy między innymi w książce R. T. Morrison, R. N. Boyd, Chemia Organiczna, PWN, W-wa, 1985, t. 1, str. 170-173.
Użytkownicy programu HyperChem mogą sprawdzić swoją odpowiedź wybierając z menu Display opcję Labels, a następnie w nowym oknie zaznaczając opcję Chirality.
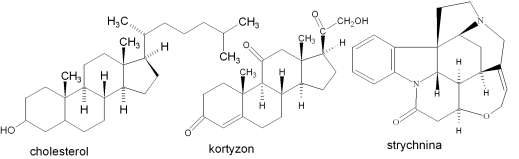
Cholesterol
6 12.127000000 -9.188000000 2.015000000
6 12.222000000 -10.488000000 1.238000000
6 9.738000000 -10.570000000 1.811000000
6 10.977000000 -11.387000000 1.386000000
6 8.469000000 -11.401000000 1.491000000
6 13.514000000 -8.621000000 1.722000000
6 10.984000000 -8.339000000 1.430000000
6 9.732000000 -9.191000000 1.105000000
6 13.562000000 -11.082000000 1.699000000
6 14.452000000 -9.828000000 1.908000000
6 11.171000000 -12.516000000 2.411000000
6 8.715000000 -12.913000000 1.798000000
6 9.814000000 -13.111000000 2.860000000
6 13.914000000 -7.438000000 2.638000000
6 11.883000000 -9.397000000 3.525000000
6 8.118000000 -11.267000000 0.000000000
6 7.302000000 -10.876000000 2.354000000
6 16.449000000 -7.552000000 2.339000000
6 15.169000000 -6.751000000 2.081000000
6 7.434000000 -13.655000000 2.244000000
6 12.806000000 -6.374000000 2.646000000
6 17.631000000 -6.774000000 1.752000000
6 5.979000000 -11.609000000 2.032000000
6 6.211000000 -13.050000000 1.544000000
1 12.298000000 -10.213000000 0.173000000
1 10.769000000 -11.831000000 0.395000000
1 9.765000000 -10.393000000 2.901000000
1 13.553000000 -8.285000000 0.666000000
6 18.959000000 -7.449000000 2.109000000
6 19.054000000 -8.840000000 1.471000000
6 20.111000000 -6.588000000 1.592000000
8 5.051000000 -13.832000000 1.780000000
1 11.306000000 -7.902000000 0.591000000
1 10.734000000 -7.637000000 2.097000000
1 8.919000000 -8.687000000 1.397000000
1 9.695000000 -9.339000000 0.117000000
1 13.944000000 -11.683000000 0.998000000
1 13.450000000 -11.592000000 2.552000000
1 14.844000000 -9.824000000 2.829000000
1 15.189000000 -9.802000000 1.232000000
1 11.647000000 -12.148000000 3.210000000
1 11.724000000 -13.238000000 1.996000000
1 9.021000000 -13.323000000 0.939000000
1 9.526000000 -12.662000000 3.706000000
1 9.928000000 -14.091000000 3.024000000
1 14.072000000 -7.796000000 3.558000000
1 12.531000000 -8.842000000 4.047000000
1 12.012000000 -10.362000000 3.753000000
1 10.949000000 -9.120000000 3.753000000
1 7.187000000 -10.912000000 -0.092000000
1 8.177000000 -12.163000000 -0.438000000
1 8.759000000 -10.636000000 -0.438000000
1 7.182000000 -9.899000000 2.177000000
1 7.524000000 -11.016000000 3.319000000
1 16.382000000 -8.447000000 1.899000000
1 16.579000000 -7.672000000 3.323000000
1 15.262000000 -5.855000000 2.514000000
1 15.060000000 -6.636000000 1.094000000
1 7.324000000 -13.566000000 3.234000000
1 7.507000000 -14.622000000 2.001000000
1 12.570000000 -6.147000000 3.591000000
1 11.998000000 -6.731000000 2.177000000
1 13.131000000 -5.553000000 2.177000000
1 17.627000000 -5.844000000 2.121000000
1 17.539000000 -6.739000000 0.757000000
1 5.496000000 -11.104000000 1.317000000
1 5.417000000 -11.638000000 2.858000000
1 6.330000000 -13.089000000 0.552000000
1 19.013000000 -7.545000000 3.103000000
1 19.101000000 -9.533000000 2.189000000
1 19.876000000 -8.890000000 0.904000000
1 18.246000000 -9.001000000 0.904000000
1 20.617000000 -6.209000000 2.367000000
1 19.745000000 -5.841000000 1.036000000
1 20.723000000 -7.150000000 1.036000000
1 4.572000000 -14.260000000 1.013000000
Kortyzon
6 10.500000000 -11.438000000 1.641000000
6 10.588000000 -12.956000000 1.425000000
6 9.206000000 -11.061000000 2.391000000
6 9.490000000 -13.736000000 1.457000000
6 7.971000000 -11.663000000 1.714000000
6 8.152000000 -13.160000000 1.694000000
6 13.041000000 -11.676000000 2.258000000
6 13.006000000 -12.516000000 0.974000000
6 11.690000000 -10.907000000 2.503000000
6 11.935000000 -13.601000000 1.163000000
6 14.058000000 -9.491000000 1.416000000
6 14.255000000 -10.725000000 2.262000000
6 13.049000000 -8.608000000 2.135000000
6 11.748000000 -9.383000000 2.265000000
6 15.468000000 -8.904000000 1.254000000
6 16.430000000 -10.072000000 1.539000000
6 15.546000000 -11.322000000 1.718000000
8 7.200000000 -13.882000000 1.886000000
6 10.483000000 -10.790000000 0.239000000
8 10.704000000 -8.776000000 2.171000000
1 11.432000000 -11.087000000 3.565000000
1 14.408000000 -10.390000000 3.304000000
1 15.593000000 -8.572000000 0.207000000
1 13.166000000 -12.387000000 3.094000000
6 13.547000000 -9.802000000 0.000000000
6 15.763000000 -7.731000000 2.169000000
8 15.181000000 -7.623000000 3.223000000
6 16.769000000 -6.686000000 1.748000000
8 17.922000000 -6.790000000 2.570000000
1 9.113000000 -10.066000000 2.403000000
1 9.259000000 -11.403000000 3.329000000
1 9.661000000 -14.710000000 1.310000000
1 7.887000000 -11.321000000 0.778000000
1 7.146000000 -11.431000000 2.230000000
1 13.898000000 -12.940000000 0.819000000
1 12.772000000 -11.937000000 0.193000000
1 11.875000000 -14.160000000 0.336000000
1 12.178000000 -14.180000000 1.941000000
1 12.894000000 -7.773000000 1.608000000
1 13.393000000 -8.371000000 3.044000000
1 16.953000000 -9.894000000 2.373000000
1 17.059000000 -10.199000000 0.771000000
1 15.387000000 -11.786000000 0.847000000
1 15.948000000 -11.965000000 2.371000000
1 10.458000000 -9.795000000 0.332000000
1 9.674000000 -11.099000000 -0.261000000
1 11.307000000 -11.057000000 -0.261000000
1 12.693000000 -10.319000000 0.060000000
1 14.231000000 -10.342000000 -0.490000000
1 13.384000000 -8.945000000 -0.490000000
1 16.390000000 -5.766000000 1.855000000
1 17.047000000 -6.824000000 0.798000000
1 18.815000000 -6.973000000 2.158000000
Strychnina
C -3.93272093 1.49162791 -1.41904651
C -4.39672093 0.18562791 -1.26604651
C -2.70772093 1.87862791 -0.88804651
C -3.64272093 -0.76337209 -0.58204651
C -1.97172093 0.92462791 -0.20904651
C -2.42572093 -0.36737209 -0.05604651
C -0.65272093 2.07962791 1.62895349
C -0.63772093 1.08762791 0.46895349
N -1.50272093 -1.16137209 0.67095349
C -0.26372093 -0.37737209 0.76795349
C 0.58927907 1.51462791 -0.23704651
C 0.83127907 1.12962791 -1.64604651
C 0.58627907 -1.02137209 -0.36604651
C 1.62727907 -0.09237209 -1.08404651
C 0.78827907 2.66062791 1.66095349
N 1.35527907 2.61062791 0.29495349
C -1.31872093 -2.63237209 0.84195349
C 1.17327907 -2.22237209 0.26895349
C 0.00227907 -3.23037209 0.19795349
C 2.70427907 0.37662791 -0.07004651
C 3.56327907 -0.51637209 0.45195349
O 2.33927907 -2.58137209 -0.46304651
C 3.55427907 -2.01637209 0.06795349
C 2.69427907 1.91062791 0.35395349
H 1.24927907 0.75262791 0.21195349
H 0.20327907 -0.46237209 1.76495349
H 1.46027907 -2.02637209 1.31895349
H 2.10427907 -0.64337209 -1.91604651
H -0.10272093 -1.35637209 -1.16504651
H -4.48572093 2.16062791 -1.91604651
H -5.28172093 -0.07437209 -1.65204651
H -2.36772093 2.81362791 -0.99004651
H -3.96672093 -1.70237209 -0.47104651
H -0.87072093 1.61162791 2.48495349
H -1.32672093 2.79962791 1.46195349
H 1.37227907 1.79262791 -2.16404651
H 0.00227907 0.90462791 -2.15604651
H 1.37127907 2.12162791 2.26895349
H 0.77727907 3.61262791 1.96695349
H -2.11672093 -3.06937209 0.42895349
H -1.31572093 -2.80637209 1.82695349
H -0.16572093 -3.45237209 -0.76304651
H 0.26827907 -4.05937209 0.68995349
H 4.18727907 -0.08137209 1.10095349
H 3.76627907 -2.56037209 0.87995349
H 4.24927907 -2.17737209 -0.63204651
H 3.01527907 1.98862791 1.29795349
H 3.30827907 2.41862791 -0.25104651
[1] W 1965 roku Robert Burns Woodward (USA) otrzymał Nagrodę Nobla za wybitne osiągnięcia w sztuce syntezy organicznej -for his outstanding achievements in the art of organic synthesis? za opracowanie między innymi syntezy cholesterolu, kortyzolu, strychniny, chlorofilu.
Strona 1 z 4