









Aktualności
Obliczanie stałych równowag reakcji dysocjacji protonu
Ćwiczenie
Obliczanie stałych równowag reakcji dysocjacji protonu
Reakcje równowag dysocjacji protonu należą do jednych z podstawowych reakcji w chemii organicznej. Najbardziej popularnymi reakcjami są reakcje z udziałem kwasów karboksylowych, bądź związków posiadający kwaśny atom wodoru. Reakcje dysocjacji możemy przedstawić za pomocą równania:
 Dla tej reakcji stała równowagi wynosi:
Dla tej reakcji stała równowagi wynosi:

Na podstawie energii swobodnej tej reakcji można określić stałą dysocjacji, która wynosi:
pKa = deltaGaq/2.303RT
Gdzie R jest uniwersalną stałą 8.314 J/(mol*K) zaś T -temperaturą (warunkach standardowych wynosi T=298.15K). Aby określić deltaG reakcji musimy użyć cyklu termodynamicznego[1] przedstawionego poniżej.
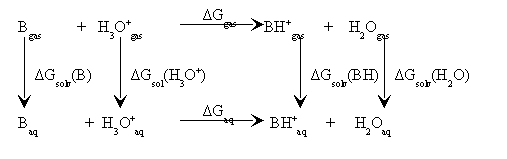
deltaG dla fazy gazowej wynosi:
deltaGgas = Ggas(H3O+) +Ggas(A-) - deltaGgas(AH) -deltaGgas(H2O)
Jeżeli obliczymy efekty związane z rozpuszczaniem (solwatacja poszczególnych składników) możemy reakcje opisać deltaG w roztworze wodnym następującym równaniem.
deltaGaq = Ggas(H3O+) +Ggas(A-) - deltaGgas(AH) - deltaGgas(H2O) +deltaGsol(H3O+) + deltaGsol(A-) -deltaGsol(AH) -deltaGsol(H2O),
GAUSSIAN
Do obliczeń pKa polecamy program Gaussian ze względu na dobre modele solwatacyjne: PCM (Polarizable Continuum Model) oraz CPCM (Conductor-like Polarizable Continuum Model). Prowadząc przedstawione poniżej obliczenia, musimy w pierwszym etapie dokonać optymalizacji struktury i obliczeń częstości. Obliczenia częstości dokonujemy dla każdego składnika osobno poprzez wybranie w programie GaussView podczas przygotowywania zadania w zakładce Job Type opcji opt+ freq. Możemy także w pliku wejściowym wpisać w linijce definiującej zadania # obok słowa kluczowego opt słowo freq. Po skończeniu obliczeń wartość Ggas określonego składnika znajdujemy w pliku z wynikami w sekcji Termochemistry w linijce:
Sum of electronic and zero-point Energies= -1910.730496
Wartość podana jest w Hartree. Przeliczając Hartree na kcal/mol mnożymy naszą wartość przez 627.5095.
Efekty solwatacyjne obliczamy zakładając, że geometria cząsteczki w rozpuszczalniku nie zmienia się znacząco oraz że w każdym przypadku dokonujemy takiego samego błędu związanego z wyżej wspomnianym założeniem, błędy te powinny się rekompensować w bilansie.2 Po zoptymalizowaniu struktury otwieramy plik programem GaussView i ustawiamy nowe obliczenia. W zakładce Job Typ wybieramy Energy, w zakładce Method wybieramy którąś z metod ab initio lub DFT. W metodach półempirycznych program nie zakłada możliwości wyboru rozpuszczalnika. Po ustawieniu tych parametrów w zakładce Solvatation wybieramy model rozpuszczalnika. Standardowym proponowanym przez nas modelem jest model CPCM. Po wybraniu modelu rozpuszczalnika wybieramy jego rodzaj rozpuszczalnika (dla obliczeń w roztworach wodnych będzie to Water).
Możemy także obliczenia przygotować poprzez dopisanie w linii zadań:
#P B3LYP/3-21g scrf=(pcm,solvent=water)
Wartości deltaGsol poszczególnych składników odczytujemy z pliku wynikowego (.log lub out) we fragmencie zawierającym informację dotyczące obliczeń zastosowaniem rozpuszczalnika. Przykład przedstawiony jest poniżej.
--------------------------------------------------------------------
Variational C-PCM results
=========================
(a.u.) = -1204.645024
(a.u.) = -1204.726009
Total free energy in solution:
with all non electrostatic terms (a.u.) = -1204.698090
--------------------------------------------------------------------
(Polarized solute)-Solvent (kcal/mol) = -50.82
--------------------------------------------------------------------
Cavitation energy (kcal/mol) = 41.48
Dispersion energy (kcal/mol) = -25.75
Repulsion energy (kcal/mol) = 1.79
Total non electrostatic (kcal/mol) = 17.52
--------------------------------------------------------------------
Suma dwóch składników, które na wydruku zostały pogrubione daje wartość deltaGsol wyrażona w kcal/mol.
HYPERCHEM
Używając program HyperChem, możemy modelować poprzez zadanie rozpuszczalnika jako modelu pudełka z wodą lub używając tylko obliczeń w fazie gazowej. W tym celu rysujemy strukturę a następnie optymalizujemy ją w pudełku z wodą poprzez wybranie z menu Setup opcji Periodic Box. Po wybraniu wymienionych opcji ustawiamy rozmiar pudełka, następnie wybieramy metodę i bazę korzystając z czterech pierwszych opcji zakładki Setup. Uruchamiamy nasze obliczenia w zakładce Compute w opcji Geometry Optimization. Energię odczytujemy z dołu ekranu, podstawiając do wzoru jako energię poszczególnych składników w roztworze Gaq. Możemy także obliczyć energię układu z zastosowaniem rozpuszczalnika. Wybieramy z menu Setup metodę Ab initio lub Density functional w oknie dialogowym, które pojawia się po wybraniu tych pozycji opcji Options a następnie w nowym oknie wybieramy Polarizability i ustawiamy parametry.
Ćwiczenie A
Z poniższej tabelki wybierz sześć związków i oblicz ich pKa używając metody B3LYP oraz HF i MP2 w bazie 6-31G(d). Korzystaj z modelu rozpuszczalnika CPCM. Porównaj wyniki z wartościami eksperymentalnymi.[3]
|
Związek |
pKa |
|
H (fenol) m-aminofenol m-bromofenol m-chlorofenol m-cyanofenol m-fluorofenol m-hydroksyfenol m-metoksyfenol m-metylofenol m-nitrofenol p-aminofenol p-bromofenol p-chlorofenol p-cyanofenol p-fluorofenol p-hydroksyfenol p-metoksyfenol p-metylofenol p-nitrofenol |
9.98 9.87 9.03 9.02 8.61 9.28 9.44 9.65 10.08 8.40 10.30 9.36 9.38 7.79 9.95 9.96 10.21 10.14 7.15 |
ĆWICZENIE B
Uszereguj poszczególne związki zgodnie z rosnącą kwasowością, korzystając z obliczeń wykonanym metodą B3LYP w bazie 6-31G(d), oraz korzystając z modelu rozpuszczalnika CPCM.
a) CH3-CH2-OH, CH3-CH2-SH, Ph-OH, Ph-SH
b) CHFCOOH, CHF2COOH, CF3COOH, CCl3COOH, CHCl2COOH, CH2ClCOOH, CH3COOH
Od czego zależy kwasowość tych związków?
*Porównaj wyniki pKa z wartościami eksperymentalnymi.[4]
Ćwiczenie C*
Korzystając z cyklu termodynamicznego na protonowanie, wyprowadź wzór na stałą Kb zasady.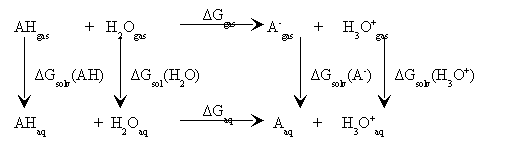 Policzy pKb dla trzech wybranych amin z zastosowaniem funkcjonału B3LYP oraz bazy funkcyjnej 6-31G(d). Porównaj wyniki z eksperymentalnymi.
Policzy pKb dla trzech wybranych amin z zastosowaniem funkcjonału B3LYP oraz bazy funkcyjnej 6-31G(d). Porównaj wyniki z eksperymentalnymi.
1 Liptak, M. D.; Shields, G. C., Accurate pKa Calculations for Carboxylic Acids Using Complete Basis Set and Gaussian-n Models Combined with CPCM Continuum Solvation Methods. J. Am. Chem. Soc. 2001, 123, (30), 7314-7319; Liptak, M. D.; Shields, G. C., Experimentation with different thermodynamic cycles used for pKa calculations on carboxylic acids using complete basis set and Gaussian-n models combined with CPCM continuum solvation methods. Int. J. Quantum. Chem. 2001, 85, (6), 727-741
2 Chcąc obliczyć geometrię cząsteczki w rozpuszczalniku dokonujemy optymalizacji cząsteczki z jednym z modeli rozpuszczalnika.
3 Kevin C. Gross, P. G. S., Substituent effects on the physical properties and pKa of phenol. International Journal of Quantum Chemistry 2001, 85, (4-5), 569-579
4 Niektóre wartości pKa znajdziesz w publikacji Benjamin G. Tehan, Edward J. L. Margaret G. W. Will R. P. John G. M. David T. M. E. G., Estimation of pKa Using Semiempirical Molecular Orbital Methods. Part 1: Application to Phenols and Carboxylic Acids. Quantitative Structure-Activity Relationships 2002, 21, (5), 457-472
Produkty reakcji mononitrowania toluenu
Ćwiczenie
Produkty reakcji mononitrowania toluenu
Jedną z najważniejszych reakcji w chemii związków aromatycznych jest reakcja substytucji elektrofilowej do pierścienia aromatycznego. W reakcji podstawienie benzenu tworzy się zwykle tylko jeden produkt. Natomiast w reakcji substytucji, w której uczestniczy wcześniej podstawiona pochodna benzenu (np.: toluenu, chlorobenzenu, fenolu) mogą powstawać trzy izomery: orto, meta i para. Czynnikiem decydującym o proporcjach produktów końcowych jest rozkład ładunku na atomach węgla i stabilności kompleksu (sigma). Energia powstawania produktów przejściowych (izomery meta lub orto, para) decyduje o końcowych produktach reakcji. Jeśli energia produktu pośredniego jest mniejsza wówczas jest większe prawdopodobieństwo powstanie tego produktu.
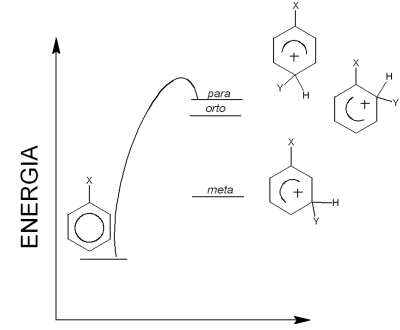
Porównując energie produktów reakcji możemy określić główny produkt, a także określić rodzaj podstawnika (I-rzędowy, II-rzędowy). Podstawniki I rodzaju powodują podstawienie w pozycji orto i para, są zwykle aktywującymi: grupy alkilowe (-CH3), hydroksylowa (-OH), aminowa (-NH2) i halogeny (-F, -Cl, -Br, -I, dezaktywują). Podstawniki II rodzaju kierują w pozycję meta i są zawsze dezaktywujące: grupa nitrowa (-NO2), karboksylowa (-COOH), amidowa (-CONH2), aldehydowa (-CHO), nitrylowa (-CN), sulfonowa (-SO3H).
Ćwiczenie A
W reakcji nitrowania możliwe jest powstanie trzech produktów przejściowych.

Na podstawie energii zoptymalizowanych struktur (B3LYP/6-31G(d)) odpowiadającym produktom przejściowym tej reakcji określ główny produkt mononitrowania toluenu. Porównaj długości wiązań C-C dla każdego produktu przejściowego w pierścieniu aromatycznym ze strukturami rezonansowymi.
Ćwiczenie B
Przeprowadź podobna analizę, lecz dla reakcji sulfonowania lub chlorowania toluenu. Porównaj wyniki z badaniami eksperymentalnymi zawartymi w literaturze.
Przestrzenna budowa związków organicznych
Ćwiczenie
Przestrzenna budowa związków organicznych
Cząsteczki związków chemicznych charakteryzują się pewną budową przestrzenną, od tej budowy zależą właściwości chemiczne i fizyczne związków. W chemii organicznej podstawowym elementem cząsteczek jest atom węgla, który może występować w hybrydyzacji sp3, sp2, sp każda z tych hybrydyzacji cechuje się odpowiednią geometrią.

- Po wybraniu pięciu cząsteczek i po z optymalizowaniu metodą Hartree Focka (HF) proszę określić strukturę przestrzenną.
- Określić na podstawie geometrii hybrydyzację poszczególnych atomów. Dla jednego przykładu pokazać drogę optymalizacji. Przygotowując obliczenia, proszę zwrócić uwagę na ładunek i multipletowość.
Kation CH5+ powstaje w fazie gazowej po przyłączeniu H+ i jest jednym z najsilniejszych -superkwasów znanych w chemii. Więcej informacji można znaleść w artukułach: D. Marx and M. Parrinello, CH5+- The Cheshire Cat Smiles, Science 284, 59-60 (1999), D. Marx and A. Savin, Topological Bifurcation Analysis: Electronic Structure of CH5+, Angew. Chem. Int. Ed. Engl. 36, 2077-2080 (1997) oraz na stronach www: http://www.theochem.ruhr-uni-bochum.de/research/marx/topic4b.en.html.
Struktura karbokationu w reakcji addycji bromu do trans-2-butenu
Ćwiczenie
Struktura karbokationu w reakcji addycji bromu do trans-2-butenu
Addycja bromu do wiązania podwójnego przebiega w dwóch etapach. Pierwszym etapem jest utworzenie stabilnego karbokationu, który przyłącza następny atom bromu tworząc wicynalną dibromopochodną. W pierwszym etapie cząsteczka Br2 zbliża się do wiązania podwójnego i następuje polaryzacja wiązania Br-Br. Następnie tworzy się bromokation. Struktura kationu będzie decydowała o produktach reakcji. Poniżej znajdują się możliwe schematy struktur karbokationów.

Strzałkami zaznaczono możliwe miejsca przyłączania drugiego atomu bromu. Dwa możliwe karbokationy, które mogą powstać będą prowadziły do różnych izomerów optycznych.
Na podstawie modelowania metodą B3LYP/6-31G(d,p) określ geometrię możliwego karbokationu. Wiedząc, że wyniku tej reakcji powstaje tylko izomer (R,S) 2,4-dibromobutanu, określ właściwą ścieżkę reakcji chemicznej i odpowiedz na pytania:
1. Dlaczego stabilniejszy jest karbokation cykliczny?
2. Jakie izomery można obserwować podczas addycji bromu do cis-2-butenu?
3. Czy wielkość atomu bromu oraz ilość jego elektronów walencyjnych ma znaczenie w tworzeniu cyklicznego kationu, porównaj swoje spostrzeżenia z innymi karbokationami (np.: CH3-CH2-CH+-CH3)
4. Jakiego rodzaju jest to addycja - syn czy anty?
Literatura
1 Roberts and G. E. Kimball, The Halogenation of Ethylenes; J. Am. Soc; 59, 947 (1937)
Molekularny elektrostatyczny potencjał -MEP
Ćwiczenie
Molekularny elektrostatyczny potencjał -MEP
- Porównaj mapy potencjałów elektrostatycznych dla anionów F-, Cl- , Br-, J-. Dlaczego mapy potencjałów różnią się, jeżeli całkowity ładunek jest taki sam dla wszystkich atomów. Obliczenia przeprowadź w metodzie B3LYP z użyciem bazy funkcyjnej 6-31G(d).
- Sporządź mapy potencjałów dla cząsteczek OH-, H3O+, H3C-CH+-CH3, NH2-, NO2+, HSO3+. Określ, do jakiej grupy czynników należą dane cząsteczki. Potwierdź to dodatkowo przykładami mechanizmów reakcji, w których uczestniczą wymienione cząsteczki. Obliczenia ? optymalizacja struktur oraz obliczenia MEP przeprowadź w metodzie B3LYP z użyciem bazy funkcyjnej 6-31G(d).
- Wykonaj mapy potencjału elektrostatycznego aminokwasów: arginina, lizyna, cysteina, metionina, walina, fenyloalanina, kwas glutaminowy, prolina, bazując na strukturach optymalizowanych metodą B3LYP w bazie funkcyjnej 6-31G(d). Czy na podstawie MEP można określić rodzaj aminokwasu aminokwasów (zasadowy, kwaśny).
- Wykonaj mapy potencjału elektrostatycznego dla uracylu, tyminy, adeniny, guaniny, cytozyny. Czy na podstawie tych obliczeń można określić komplementarność zasad? Obliczenia przeprowadź w standardowej metodzie B3LYP z użyciem bazy funkcyjnej 6-31G(d).
Obrót protonów grupy metylowej w projektowaniu nano-zabawek
Ćwiczenie
Obrót protonów grupy metylowej w projektowaniu nano-zabawek
Polarność rozpuszczalników
Ćwiczenie
Polarność rozpuszczalników
Nie jednokrotnie na zajęciach laboratoryjnych z chemii organicznej wykorzystywane są dwa typy rozpuszczalników polarne i nie polarne. Rozpuszczalniki stosowane to:
Tautomeryzacja poprzez wiązanie wodorowe w N-salicylidenoanilinie
Ćwiczenie
Tautomeryzacja poprzez wiązanie wodorowe w N-salicylidenoanilinie
Ciekawym przykładem reakcji jest tautomeryzacja N-salicylidenoaniliny, która wykazuje właściwości fotochromowe. W stanie podstawowym w ciemności substancja ta jest żółta, a po naświetleniu ultrafioletem (czyli po dostarczeniu energii do układu) przybiera barwę czerwoną. Zmiana barwy jest wyniku powstania formy tautomerycznej, która jest odpowiedzialna za fotochromizm.
Wiązania wodorowe a temperatura wrzenia
Ćwiczenie
Wiązania wodorowe a temperatura wrzenia
Temperatura wrzenia wody pod ciśnieniem 1013hPa wynosi 100°C, natomiast siarkowodoru -62.2. Na modelowym układzie ? dimer wody oraz dimer siarkowodoru oblicz energię dysocjacji dimerów, energie oddziaływania oraz błąd superpozycji bazy. Obliczenia przeprowadz metoda MP2/6-31G(d).
Energia obrotu grupy metylowej -CH3
Ćwiczenie
Energia obrotu grupy metylowej
Rotacja protonów w grupie metylowej -CH3 w pochodnych benzenu wymaga niewielkiej ilości energii. Energia ta zmierzona eksperymentalnie dla m-florotoluenu wynosi 4.39, dla o-florotoluenu 65.26, p-florotoluenu 13.82 oraz toluenu 13.94 kcal/mol.[1]
Geometria wiązań wodorowych
Ćwiczenie
Geometria wiązań wodorowych
Wiązania wodorowe (ang. hydrogen bond) możemy podzielić ze względu na liczbę geometrię oraz ilość oddziaływań, w których dany proton uczestniczy jako liniowe najprostsze, oraz osiowe (ang. bifurcate) jako bardziej złożone.
Komplementarność zasad purynowych
Ćwiczenie
Komplementarność zasad purynowych
Zasady azotowe: adenina (A), cytozyna (C), guanina (G), tymina (T) uracyl (U) w chodzącą w skład nukleozydów, które są połączeniem tych zasad z rybozą, deoksyrybozą lub rybitolem poprzez wiązanie N-glikozydowe. Estry nukleozydów z kwasem fosforowym (V) tworzą nukleotydy które są monomerami kwasów rybonukleinowych DNA i RNA. DNA jest nośnikiem informacji oraz występuje w chromosomach. Cząsteczka DNA przeważnie zbudowana jest z dwóch łańcuchów, które biegną antyrównolegle (tzn. koniec jednego łancucha jest na przeciw początku drugiego). Łańcuchy kwasu nukleinowego owijają się wokół wspólnej osi tworząc prawoskrętną podwójną helisę. Reszty fosforowe i cukrowe znajdują się na zewnątrz helisy i połączone ze sobą poprzez wiązanie fosfodiestrowe. Zasady skierowane są do wnętrza i tworzą pary zasad. Połączenie zasad jest poprzez wiązania wodorowe odpowiednio: A-T (A-U) G-C T-A (U-A) C-G.
Widmo podczerwieni i Ramana dla acetonu, tiomocznika i kwasu malonowego
. Przedstawiamy przykład obliczeń widm IR, Ramana dla acetonu, tiomocznika, i kwasu malonowego, oraz porównanie wyników teoretycznych z badaniami eksperymentalnymi. Autorką badań jest pani mgr inż. Karolina Gluza z Politechniki Wrocławskiej...
Przedstawiamy przykład obliczeń widm IR, Ramana dla acetonu, tiomocznika, i kwasu malonowego, oraz porównanie wyników teoretycznych z badaniami eksperymentalnymi. Autorką badań jest pani mgr inż. Karolina Gluza z Politechniki Wrocławskiej...
Gaussian - podstawowe informacje
To "klasyka" kwantowej chemii obliczeniowej, efekt pracy kilku pokoleń teoretyków, pierwotnie skupiających się wokół Johna Popla, który w 1998 roku otrzymał wspólnie z Walterem Kohnem nagrodę Nobla za wkład w metody obliczeniowe w chemii.
Programem tym można obliczyć wiele właściwości molekuł i reakcji, włączając w to:
- energie i strukturę cząsteczek,
- częstości drgań,
- widma IR, Ramana i UV-Vis,
- własności termochemiczne,
ArgusLab - Rafał Koczeń

ArgusLab jest programem bezpłatnym, oferuje on użytkownikowi możliwość tworzenia struktur chemicznych, umożliwia także wykonanie obliczeń optymalizacji geometrii i własności molekularnych z wykorzystaniem metod półempirycznych MNDO, AM1, PM3 i ZINDO...
Strona 3 z 4